Review of Methods for Estimating the Prevalence of Rare Diseases

Abstract
One of the main challenges in rare diseases is the unavailability of reliable estimates of prevalence and incidence. The lack of epidemiological data makes planning for therapeutic and management options challenging. Methods for estimating the prevalence and incidence of rare and genetic diseases primarily rely on the availability of accurate national patient registries or databases of birth defects. This gap is wider in Low- and Middle-Income countries (LMICs) such as India, where currently, the estimates of prevalence and incidence are either unknown or data from developed countries have to be used as a proxy. Here, we analyzed the current methods used to estimate the prevalence and incidence of rare genetic diseases to provide recommendations in the form of a decision tree to select the most feasible method, particularly in resource-constrained environments such as India. We selected ten rare diseases of shared importance to the Indo US Organization for Rare Diseases (IndoUSrare) and its Patients Alliance members for analysis. Our analysis suggests that retrospective study designs are the most commonly used method to estimate the prevalence and incidence of rare diseases. We propose a generalized decision tree or flowchart to aid epidemiology researchers during the selection of methods for estimating the prevalence and incidence of a rare or genetic disease.
Keywords
Rare disease, prevalence, incidence, epidemiology, genetic disease
INTRODUCTION
Rare diseases as a group are heterogeneous. They range from congenital malformations, developmental disorders, and autoimmune diseases to rare cancers and infectious diseases. The majority of rare diseases (about 80%) are genetic in origin, affecting 3%-4% of births, and begin in childhood[1,2]. Overall, there are estimated to be 5,000-7,000 rare diseases depending on the source, with a recent study estimating this number to be 10,867[3]. Though individually rare, these diseases collectively affect 3.5%-5.9% of the world’s population – approximately 400 million people worldwide[4,5]. The total number of affected individuals, taken together with the “high medical burden to individual patients, families, and health care systems”[6], reveals the true impact of rare diseases.
Rare diseases lead to significant healthcare spending irrespective of a country’s size and demography[6]. They need multidisciplinary resource-intensive care, the need for which increases with disease progression[7]. For individuals and societies, these are direct medical costs, costs related to informal care, and due to loss of productivity. The impact on families is often catastrophic in terms of emotional as well as financial strain, as the cost of treatment is often prohibitively high[7-10]. For many countries, the limited resources jeopardize resource distribution and the opportunity cost of investing in rare disease research and treatment in the face of other imminent disease problems that can be effectively managed with a fraction of the costs of rare diseases. Rare diseases are often chronic, terminal, disabling, and limiting, and require prolonged specialized care. They disproportionately impact children: 50% of new cases are in children, rare diseases are responsible for 35% of deaths before the age of 1 year, 10% between the ages of 1 and 5 years, and 12% between 5 and 15 years[11].
BACKGROUND
Even as new diseases are being added to the list, there is no universal consensus on what defines a rare disease. A systematic review identified 296 definitions for rare diseases from 1,109 organizations[12]. Among others, a rare disease has been defined on different occasions as a disease with an average global prevalence of 40-50 cases per 100,000 people[12], any disease affecting less than 200,000 people in the USA[13], and as a life-threatening or chronically debilitating condition affecting no more than 1 in 2,000 people in the European Union[14]. Incidence is the rate of new cases of a disease occurring in a specific population over a defined period of time. Prevalence is a measure of the total number of people affected by a disease at a given point or period of time[15]. Prevalence and incidence are important tools that guide policies and public health measures. Prevalence has been used to define the “rareness” of a disease. In the Indian public health context, there is still no formal national definition of what constitutes a rare disease. The recently announced National Policy for Rare Diseases (NPRD) identified only a small list of diseases under its ambit and did not offer a generalized definition[11]. However, medical geneticists and patient advocacy groups have proposed to set the definition to any disease affecting 1 in 5,000 individuals in India[16]. Additionally, regulators in India have defined an orphan drug as any drug that is intended to treat a medical condition affecting fewer than 500,000 individuals in the country[17]. This highlights the inconsistency and confusion surrounding the definition of rare diseases across countries and organizations. Table 1 provides a summary of a few of these definitions from across the world.
Table 1 - Varying definitions of a rare disease from different countries and regions
Organization/Country | Prevalence per 100,000 |
|---|---|
ISPOR Rare Disease Special Interest Group | 40-50[12] |
EU | 50[14] |
USA | 60[13]* |
BRAZIL | 65[18] |
Japan | 40[19] |
Canada | 50[20] |
South Korea | 40[21] |
Taiwan | 10[22] |
* Calculated value as per population in 2023[23]. In the United States, rare diseases are defined as any condition affecting less than 200,000 people in the country.
Unfortunately, existing methodologies are not sufficient to acquire prevalence data for rare diseases. While the problem of measuring prevalence persists in all countries, the issue is particularly aggravated in LMICs, which suffer because of resource-constrained health infrastructure and a lack of universal health coverage, national patient registries, or newborn screening programs[24-26]. Rare diseases pose unique challenges, beginning with uncertainty around estimates of people affected, i.e., prevalence and incidence. These challenges also include the rarity of the disease, the ability of the healthcare systems to properly diagnose a rare disease, logistics involved in and access to diagnostic facilities for geographically dispersed patients, and a shortage of medical education in handling rare diseases. For any disease, a fundamental step for garnering adequate resources for the discovery and development of novel treatment options, and payor or government reimbursement schemes for patients, is to correctly estimate the prevalence and incidence in a given region. Prevalence and incidence estimates are available only for a few relatively more common rare conditions, such as Duchenne Muscular Dystrophy[7] and Spinal Muscular Atrophy[27]. Rare diseases that have been more recently identified, e.g., Sodium voltage-gated channel alpha subunit 8 related Epilepsy (SCN8A epilepsy), or Okur-Chung neurodevelopmental syndrome (CSNK2A1 or OCNS), lack any prevalence or incidence estimates[28-30]. There is an absence of reliable information on the prevalence and incidence of most rare diseases at the national and global levels[31]. Our analysis shows that currently available literature on these estimates is based mostly on records maintained in tertiary care centers or databases. Such sources of information have inherent limitations such as selection bias (Only most severe cases and patients with a certain spending capacity reach tertiary care centers or avail insurance)[32]. Additionally, most available literature on prevalence and incidence comes from higher-income countries. Low and Middle-Income Countries (LMICs) do not have adequate prevalence and incidence estimates. This can be attributed to the lack of robust methods to estimate the incidence and prevalence of rare diseases in LMICs.
Robust estimates for the prevalence and incidence of rare diseases are necessary to develop public health policy frameworks, prioritize further research towards prevention, resource planning, regulatory pathways for orphan drug development, develop Governmental special incentive programs, and early and adequate treatment that optimizes the quality of life for the affected people. Without prevalence or incidence data, planning for therapeutic and management options becomes a challenge. Policymakers need disease-specific prevalence and related epidemiologic burden indicators to decide on appropriate resource allocation; regulatory agencies rely on these numbers to provide “orphan’’ designation for drugs, which in turn is essential to support orphan drug discovery and development. Governments have the mandate to ensure equitable access to healthcare for all citizens, especially for medically underserved populations.
There is very limited literature focusing on rare diseases in India. It is estimated that India has around 70 million people affected by rare diseases[33]. Rare diseases that have come into the policy discourse in India are blood disorders, lysosomal storage diseases, primary immunodeficiency diseases, mitochondrial diseases, neurodegenerative diseases, and musculoskeletal diseases[34]. India has a high and prevalence of birth defects, hemoglobinopathies, inborn errors of metabolism, and Down’s syndrome[35-40]. Many of these birth defects are rare diseases and are underestimated in prevalence, thereby contributing to a huge economic and social burden[41]. Here, we undertook a scoping review to analyze the current gaps in methods used to estimate the prevalence and incidence of rare diseases to provide recommendations to strengthen epidemiological approaches to rare diseases, particularly in LMICs such as India.
METHODS
Identification of diseases for analysis
To study and analyze the epidemiological methods used for estimating the prevalence and incidence of rare diseases, we selected ten rare diseases of shared importance to the Indo US Organization for Rare Diseases (IndoUSrare)[42] and its Patients Alliance members. These ten diseases are Spinal Muscular Atrophy (SMA), Duchenne Muscular Dystrophy (DMD), Ehlers-Danlos Syndrome (EDS), Pemphigus and Pemphigoid, Okur-Chung Neurodevelopmental Syndrome (CSNK2a1/OCNS), SCN8A epilepsy, Mucopolysaccharidoss type II (MPS type II), Gaucher Disease, and X-linked Adrenoleukodystrophy (X-linked ALD). These ten diseases represent the spectrum of rare diseases, from the commonest DMD (Prevalence 7.1 per 100,000; Incidence 19.8 per 100,000) [7] to extremely rare like Okur Chung Neurodevelopmental syndrome (OCNS with only 120 diagnosed patients worldwide)[30], and SCN8A epilepsy (with around 450 reported cases globally)[29].
Information sources and search strategy
We surveyed the scientific literature to identify previously published original research articles, reviews, and systematic reviews on the epidemiology of these diseases. Due to the extensive nature of this review and issues related to the accessibility of literature, the searches were limited to PubMed. PubMed enlists articles that are peer-reviewed and from indexed journals, thereby ensuring maximum quality. The search terms used were identified after consultation among the authors and an initial review of rare disease literature. The following terms were used in combination to identify relevant articles on PubMed: “incidence”, “prevalence”, “epidemiology”, “Rare disease”, and “Genetic disease”, along with specific disease names (“Spinal Muscular Atrophy”, “Duchenne Muscular Dystrophy”, “Ehlers Danlos Syndrome”, “Pemphigus and Pemphigoid”, “Okur-Chung Neurodevelopmental syndrome (CSNK2a1)”, “SCN8A epilepsy”, “Mucopolysaccharidoss type II”, “Gaucher Disease” and “X-linked Adrenoleukodystrophy”). Searches were conducted between July – September 2021.
Eligibility criteria
Papers not available in English and not pertaining to humans were excluded through search filters for language (English) and species (Humans), with no time limitations set. Initially, a general search was conducted to identify reviews or original articles that discussed the incidence and prevalence of rare or genetic disorders, without adding restrictions through disease-specific terms. Since the initial search for broad categories of rare and genetic diseases was to determine the overall landscape and understanding of the epidemiology of rare diseases and to identify search terms for use in this study, the analysis was limited to a screening of the most recent 400 articles in the search results.
Disease-specific searches were then performed to identify articles providing insights into the incidence or prevalence of each of the ten selected diseases. Individual searches were run for all diseases except Pemphigus and Pemphigoid, which were combined. Results from such disease-specific literature searches were screened completely to identify original articles and reviews providing information on the prevalence and incidence of the particular rare disease. The identified articles were then retrieved for full-text screening. Relevant information was extracted from the original articles and systematic reviews using a structured spreadsheet [Supplementary Table 1] into the following columns: study ID, country, study setting, study population, study design, duration of the study, methods used, objectives of the study, incidence, prevalence, other findings, and remarks or conclusions. References within selected articles were cross-checked to locate further articles of relevance that might have been missed during the screening of search results. The articles identified by cross-referencing were evaluated and relevant information was extracted as described above. Wherever recent systematic reviews and meta-analyses that dealt with prevalence and incidence estimates for diseases of interest were available, individual studies for those diseases were not utilized to create the epidemiology summary due to extensive coverage in systematic reviews.
Data analysis
The data extracted as described were analyzed and cross-referenced with information available on websites that focused on specific diseases or rare diseases in general, to obtain an overview of the current epidemiological knowledge for the selected diseases. The data are presented in Table 2 and include information on inheritance, treatment options, and prognosis.
Construction of decision tree
Based on the inferences drawn from the analysis of studies reviewed, a decision tree was drawn to aid in the identification of appropriate methods for use in the future to study rare diseases with consideration for prevalence burdens, settings, and resources. The decision tree was constructed based on an analysis of the methods used for rare diseases and the type of data collected in the reviewed studies. It was further modified and restructured to include steps to estimate the prevalence based on meetings and discussions among authors.
RESULTS
Our searches resulted in the identification of the latest comprehensive studies on the ten selected rare diseases. The number of articles analyzed for each disease in the preliminary search and those obtained post-screening are summarized in Table 2.
Disease name | Number of articles preliminary search | Number of articles post screening | |
|---|---|---|---|
Original articles | Systematic /scoping reviews | ||
SMA | 900 | 8 | 1 |
DMD | 1,008 | 20 | 2 |
EDS | 469 | 3 | 0 |
Pemphigoid | 1,547# | 11 | 2 |
Pemphigus | 28 | 3 | |
Okur Chung neurodevelopmental syndrome | 21*± | 0 | 0 |
SCN8A epilepsy | 488$* | 4 | 0 |
MPS II | 182 | 13 | 0 |
Gaucher disease | 841 | 4 | 1 |
X-linked ALD | 186 | 10 | 0 |
Characteristics of the rare diseases studied
The ten diseases analyzed in this study reflect the diversity seen among rare diseases in general. Eight out of ten are rare genetic diseases – All 8 genetic diseases have identified genetic biomarkers; two of these were recently identified: SCN8A epilepsy (2012)[43] and OCNS (2016)[44]. Pemphigus and Pemphigoid are multifactorial with unknown genetic biomarkers. DMD, MPS II, ALD, and EDS subtypes are X-linked recessive and, thus, very rarely affect females. Penetrance for genetic diseases is mostly variable. The methods of diagnosis include genetic, biochemical, histological, and the identification of specific sets of clinical signs and symptoms. Most of the rare diseases analyzed manifest early in life, except for Pemphigus and Pemphigoid, for which symptoms start at an older age. All diseases studied shorten life expectancy. Patients suffering from the rare diseases analyzed require lifelong supportive therapy, with some being responsive to therapeutic modalities such as enzyme therapy, gene therapy, and bone marrow transplantation. Most diseases have an assigned ICD (International Classification of Diseases) 10[45] or ICD 11 code[46], except for recent additions like SCN8A and OCNS. Table 3 summarizes the current knowledge about each of the diseases considered for this study.
Disease | Inheritance | Penetrance | Prevalence | Diagnosis | Treatment | Prognosis | Codes |
|---|---|---|---|---|---|---|---|
Incidence | |||||||
Duchenne Muscular Dystrophy (DMD) | X-linked Recessive, 1/3rd mutations are De novo and 2/3rds Inherited, > 150 mutations in DMD gene (Xp21.2), Extremely rare cases of females also affected | Variable penetrance | Prevalence 7.1 per 100,000[7] | Symptoms, Clinical Examination, elevated creatine kinase blood levels, Muscle Biopsy (Dystrophin protein levels), Genetic Testing (Gold Standard) | Symptom management with steroids, additional specific medication, and assistive devices available. | Age of onset: Childhood. Symptom onset before 6 years of age, progressive, shortened lifespan, rarely live beyond the 20s-40s. | ICD-10: G71.0 |
Incidence 19.8 per 100,000[7] | |||||||
Spinal muscular atrophy/ Proximal spinal muscular atrophy | Multiple subtypes, Autosomal Recessive, Mutations (Homozygous deletions) in SMN1 gene (5q), and SMN2 gene (modifies severity). Only 2% de novo mutations. > 70-110 mutations known | Variable penetrance | Prevalence: 1-2 per 100,000[27] | Symptoms, Electromyography, and muscle biopsy, and Genetic testing (Confirmatory) | Supportive (nutritional, and physical therapy) | Age of onset: All ages, Variable; Symptom onset can range from during pregnancy to childhood; on specific subtypes, earlier-onset forms are generally associated with a poor prognosis | ICD-10: G12.0 G12.1 |
Incidence 10 per 100,000 live births[27] | |||||||
X-linked Adrenoleukodystrophy | X-linked (Xq28), ABCD1 gene (95% inherited from parent, 4% de novo) | 100 % penetrance in males; | Prevalence: 0.8 per 100,000[47] | Testing levels of very long-chain fatty acids (VLCFA), C26:0-lysophosphatidylcholine in , functional assays in cultured skin fibroblasts, Brain MRI, | Corticosteroids, Physical therapy, Lorenzo’s oil (Experimental), Bone marrow transplantation [Allogeneic hematopoietic stem cell transplantation (HSCT)] | Age of onset: All ages. Three main types out of 8 symptom subtypes. 20% of Carrier Females also show symptoms. Variable prognosis depending on symptom subset | ICD 10: E71.3 Disorders of fatty-acid metabolism |
Incidence: 0.8[48] -20.6[49] per 100,000 | |||||||
Gaucher disease | Lysosomal storage disorder, three main forms (types 1, 2, and 3), Autosomal Recessive, GBA gene (1q21, 1q22) | High penetrance in Homozygotes | Prevalence: 0.7-1.75 per 100,000 | Chemical analysis (thin-layer chromatography and gas-liquid chromatography) of the sediment from a 24-h urine collection, assay of leukocyte beta-glucosidase. Genetic testing (confirmatory) | Enzyme replacement therapy, Chemical Chaperone Therapy, and substrate reduction therapy. These treatments are ineffective for GD type 2. | Age of onset: All ages. Prognosis variable depending on type and subtype | ICD-10: E75.2 Other Sphingolipidoss |
Incidence: 0.39-5.8 per 100,000[50] | |||||||
Mucopolysaccharidosis type 2/ Hunter syndrome/ | X-linked recessive, IDS gene (Xq28; gene encoding iduronate 2-sulfatase). Rarely female carriers affected | 100% in males | Prevalence: 0.07[51] - 7.06[52] per 100,000 | Clinical s/s, Elevated dermatan sulfate (DS) and heparan sulfate (HS) in urine, iduronate-2-sulfatase (I2S) enzyme deficiency in the serum, leukocytes or fibroblasts, or in dried blood spot samples Genetic testing | Enzyme replacement therapy and Extensive palliative care | Age of onset: Childhood Symptoms begin between 18 months to 4 years of age | ICD-10: E76.1 ICD-11: 5C56.31 |
Incidence: 0.26[51]- 3.08[52] per 100,000 | |||||||
Ehlers Danlos Syndrome | Group (Group A-F) of related disorders caused by different genetic defects in collagen | Prevalence: 0.2 % or 200 per 100,000[53] | History and clinical Examination, Electron microscopy. Skin biopsy and genetic testing | Treatment directed at preventing complications and symptom management | Age of onset: Infancy, Neonatal. Prognosis variable, vascular type is severe with limited lifespan | ICD-10: Q79.6 | |
Incidence of all EDS types: 1/ 2,500 to 1/5,000 births* | |||||||
SCN8A epilepsy | Autosomal Dominant (12q13.13), De novo mutations mostly, ~450 patients worldwide | "Unknown but assumed to be complete | 1% of all cases of epileptic encephalopathy* | History and genetic testing | Treatment aimed at seizure control using medication | Age of onset: 0-18 months, at a mean age of 5 months | |
Incidence: 0.6[54] - 2.75[55] = per 100,000 | |||||||
Okur-Chung Neurodevelopmental Syndrome | Autosomal Dominant, 20p13 (CSNK2A1), Heterozygous mutations- de novo mutations | 100% affected with variable phenotypic expression | > 120 patients worldwide*, Prevalence/Incidence unknown, | Whole exome sequencing | Supportive therapy | Mostly unknown, still being explored. Age of onset: Childhood | |
Pemphigus | Group of rare autoimmune diseases | Prevalence: 6-14.8 per 100,000[56] | Testing based on clinical, histological, and direct immunofluorescence methods | Treatment aimed at symptomatic relief using steroids, immune modulators, and antibiotics | Age of onset: at any age, common in middle or old age. Prognosis variable, depending on the subtype, can be life-threatening | ICD-10: L10 | |
Incidence: 0.5-16.1 per 100,000[56] | |||||||
Pemphigoid | Group of subepidermal, blistering autoimmune diseases | Prevalence estimates: 0.01 per 100,000[57] | Testing based on clinical, histological, and direct immunofluorescence methods | Treatment aimed at symptomatic relief using steroids, immune modulators, and antibiotics | Bullous Pemphigoid Age of onset: elderly or old age (> 65yrs) | ICD-10: L11 | |
Incidence: 0.48-1.37 per 100,000[58] |
DMD
In a systematic review and meta-analysis published in 2020, the estimated prevalence of DMD among males is 7.1 per 100,000 males (95%CI: 5-10.1), with a birth prevalence of 19.8 per 100,000 live male births (95%CI: 16.6-23.6). The overall population prevalence was estimated as 2.8 cases per 100,000 (95%CI: 1.6-4.6). The authors estimated very high between-study heterogeneity, which can be attributed to methodological differences in studies, and the majority of the studies were found to be of medium quality based on an algorithm that took into account a number of factors including descriptions of study design, eligibility criteria, study population, and outcomes[7]. In another systematic review of 10 population-based studies on DMD, the authors reported a prevalence that ranges from 0.95 per 100,000 in South Africa to 16.76 per 100,000 in Sweden[59]. The review also reported the incidence rate of DMD in the Canadian population as 1 per 3,600 (or 27.8 per 100,000) live-born males per year. The incidence rate in European countries ranged from 10.71 (Italy) to 18.8 (Denmark) per 100,000 live-born males per year. The overall pooled estimate of the prevalence of DMD among males worldwide was 4.78 (95%CI 1.94-11.81) per 100,000.
SMA
For SMA, eight original articles and one systematic review were analyzed. The systematic review reported a prevalence of approximately 1-2 per 100,000 persons and an incidence of around 1 in 10,000 live births has been estimated, with SMA type I accounting for around 60% of all cases[27]. The authors also highlight that most of the studies were old and relied mostly on clinical diagnosis, performed in a small geographic area, mostly in European populations.
OCNS
Okur Chung Neurodevelopmental Syndrome (OCNS) is a very rare disease described in 2016[44] with around 450 patients[30] identified until now. OCNS has 35 CSNK2A1 variants in various protein-coding regions of CK2α. Due to the rarity of the condition, the genotypic and phenotypic variations have not been well understood till now. We could find only case reports and case series on OCNS [Supplementary Table 1], none of which reported the prevalence or incidence of OCNS.
SCN8A epilepsy
SCN8A epilepsy is another relatively newly identified genetic disorder causing epilepsy[43]. Four full texts that met our criteria were reviewed, of which two were clinic-based studies focusing on genetics and inheritance without any information on incidence and prevalence[60,61]. One study[54] conducted a prospective epidemiological study in Scotland and reported an estimated incidence of 0.6 per 100,000 live births, while the other[55] showed an estimated incidence of 2.75 per 100,000 live births in an epidemiological study conducted in Tasmania from 2011 to 2016.
Gaucher Disease
Four full-text original articles were identified for Gaucher disease, three of them reported incidence, and none reported prevalence[62-65] .A systematic review that summarized findings from forty-nine studies reported a standardized birth incidence of Gaucher disease in the range of 0.39 to 5.8 per 100,000 based on 11 studies and prevalence in the range of 0.70-1.75 per 100,000 based on 9 studies[50].
MPS II
A total of 13 original articles were reviewed for Mucopolysaccharidosis type II from screening search results and cross-referencing. Our search yielded no systematic reviews or reports. The incidence of the disease ranged from 0.26 per 100,000 in the USA[51] to 0.43 per 100,000 in the Czech Republic[63], up to 3.08 per 100,000 in Norway[52]. The prevalence was reported as 0.7 per 1,000,000[51] up to 7.06 per 100,000[52].
EDS
Three original articles that report the prevalence of EDS were found, and none were found that reported incidence. The reported prevalence ranged from 0.02% to 0.0002%. The report identified a low national prevalence of patients diagnosed with EDS in Denmark and showed that the majority of patients diagnosed are female[53]. The EDS cohort had a lower educational level, mean age, and life expectancy than the background population, and showed a predisposition for receiving state-granted subsidies.
X-linked ALD
Ten original articles were reviewed for X-linked ALD. Only one reported a prevalence of 0.8 per 100,000 in Norway[47]. The reported incidences varied between studies. This range included 0.8 per 100,000 in Germany[48], 1 per 100,000 in France[66], and 20 per 100,000 in the United States[49].
Pemphigus and Pemphigoid
Pemphigus has been reported to have an incidence of 0.5-16.1 per 1,000,000 and a prevalence of 60-148 per 1,000,000[56]. Based on their analysis of 38 studies published between 1952 and 2015, the authors of the study note an increasing trend in the incidence in recent years. A recent systematic review report on Bullous Pemphigoid reviewed 26 observational studies to report a global incidence of 8.2 per 1,000,000, ranging from 4.8 to 13.7 per 1,000,000[58].
An analysis of the methods used and a roadmap for epidemiological studies in rare diseases
Most full-text articles reviewed for this study have used a retrospective study design that accumulates data from laboratory or clinic-based records and less frequently from disease-specific databases, national health directories, and prospective or cross-sectional studies. Additionally, the systematic reviews that have been appraised for this study agree on the heterogeneity and moderate-poor quality of the articles therein[7,27,50,56,58,59,67].
The prevalence estimates obtained and shown in Table 2 are based on those reported in the studies analyzed. Most of these corelate with the ranges state in the Orphanet Database. Only two diseases have values not comparable with the data reported from Orphanet (Pemphigoid and X-linked ALD). This can be attributed to the stringent criteria followed by Orphanet for inclusion of studies, and that the data reported in these cases are from Europe only. Two of the diseases do not yet have an ORPHA code – SCN8A epilepsy and OCNS.
Based on our analysis of various studies described in this article, a decision tree was designed, which provides a roadmap that investigators can follow to best utilize available knowledge, resources, and design methodology to arrive at the optimal method for estimating the prevalence of a rare disease. This can be used as an aid to decide upon the most suitable study design and methodology for epidemiological studies for any rare disease, taking into account the status of current research and knowledge available for the particular disease. The proposed decision tree is described as a flowchart in Figure 1. The flow chart is non-exhaustive, with the intent of facilitating the choice of an appropriate method(s) for the estimation of the prevalence of any rare disease.
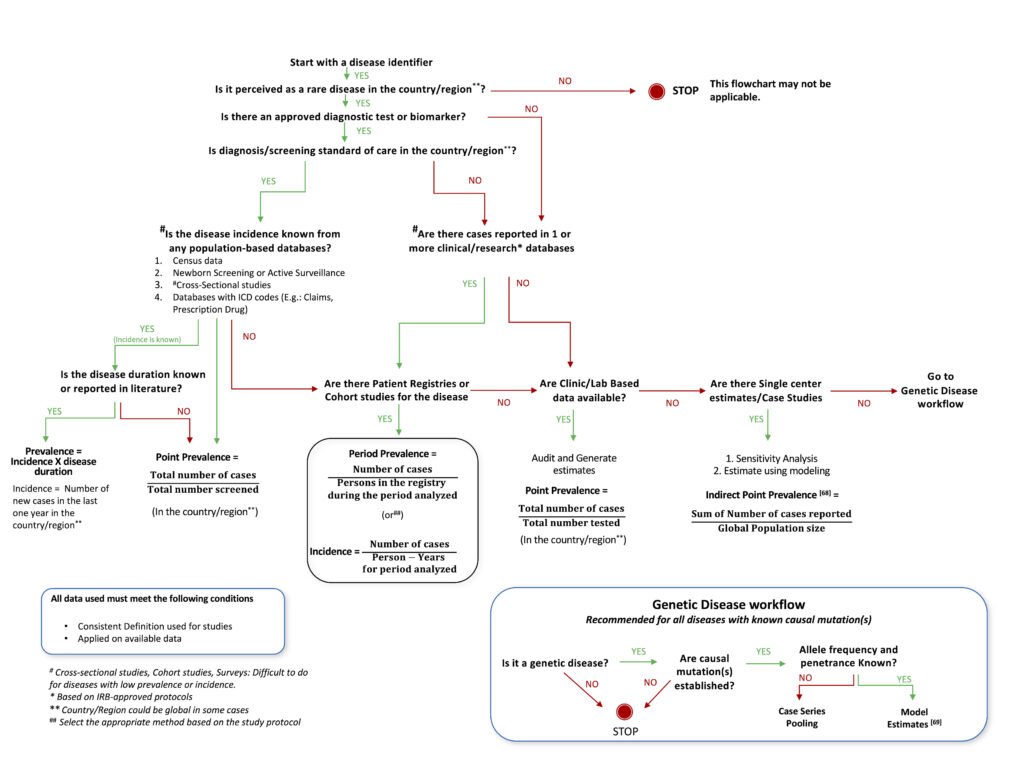
Epidemiology researchers could consider the flow chart while choosing methods to estimate the prevalence of rare diseases. The proposed decision tree utilizes traditional well-established epidemiological methods such as cross-sectional, cohort studies, case studies[68] or survey-based analysis and proposes modeling through available genetic data[69] as an alternate, and possibly only method because traditional methods may prove difficult in rare conditions with a small affected population. This has immense potential for generalizing since 80% of rare diseases are thought to be genetic in origin. Geneticists should also take into account haplotypes and haplogroups that are race/ethnicity-dependent, in this method of estimation.
DISCUSSION
Even though some literature is available for rare diseases that are more common and widely studied, heterogeneity in study methods is a hindrance to synthesizing summarized evidence[7,27,50,58]. Furthermore, the limited amount of available data on rare diseases is usually from studies based in European and North American countries, mostly from Caucasian populations[7,27,50,56,70]. A recent systematic review and meta-analysis on the global epidemiology of DMD enumerated the studies country-wise and found that almost all studies were from higher-income countries, i.e., the Global North[7]. It is important to have prevalence estimates and detailed natural histories from all regions in order to better understand the differences in symptoms, prognosis, and treatment effects in the general population.
The quality of the available data is also mediocre. The limited hospital-level data from regions of higher prevalence, and the variations in contexts and definitions, make such estimates not fit for generalization at a global level. The absence of local evidence leads to becoming dependent on evidence originating from elsewhere and the disease(s) not being prioritized as a public health issue. In cases without estimates, cases or families documented in medical literature are the only sources providing some insight. The majority of literature on rare diseases is in the form of case reports in different languages and published several years apart[71-74]. Case reports and series do not help in providing accurate prevalence estimates. This leads to a situation where it is difficult to define the disease or find resources to estimate prevalence.
The definition of what constitutes a rare disease is also limited by the context, such as geographical disparities, ethnic groups, different national or organizational terminologies, and diagnostic capacities. These varied and inconsistent definitions are a roadblock to framing policy and collaborating for rare disease research. International consensus in rare disease definition and global collaboration using recent genetic diagnostic and treatment advances is a prerequisite for coordinated and organized efforts. Financial support, insurance coverage, and reimbursement rules differ between countries and regions, but access to these schemes gets progressively more difficult when the required treatment is more expensive[75]. Policies specific to rare diseases are very important in this scenario due to the potential impact of rare diseases and their treatment on the health budget of the country. In India, as per the National Policy for Rare Diseases, prevention of the occurrence of rare diseases is the main focus with an aim to limit disease burden. The policy also states that rare disease patients are entitled to financial support of up to 5 million Indian Rupees per patient for treatment of the rare diseases listed in the policy[76]. However, the policy recognizes only a limited number of rare diseases[11] and leaves many patients struggling to access benefits from government aid.
Limitations of traditional epidemiology methods for studying rare diseases
Traditional methods of estimating the prevalence of a disease include surveys, cross-sectional studies in large populations, and cohort studies with longer follow-up periods. Such study designs are resource-intensive, even for diseases that are not rare. Rare diseases pose extra difficulties – the lower number of cases and the geographically dispersed distribution of patients make these traditional methods impractical. Epidemiological studies on rare diseases are made challenging due to the heterogeneity in different rare diseases and among the patients with the same rare disease, such as the variations in etiology (genetic, environmental, infectious), the wide spectrum of phenotypic expression of disease, severity of symptoms, and possibilities of variation in genotype-phenotype correlation. Hence, the skills and training required to diagnose a rare disease are more intensive, and logistics like laboratory capacity need to be better.
The NPRD also recognizes the importance of prenatal and newborn screening as the best and most cost-effective way to manage the burden of rare diseases. A recent study explored the utility of India’s comprehensive newborn screening program – the Rashtriya Bal Suraksha Karyakram (RBSK) towards the implementation of NPRD guidelines. RBSK encourages a combined approach that uses different modes of screening in a tiered manner to ensure the identification of a wide range of diseases[77]. This involves low-cost methods like physical screening for visible birth defects to biochemical analysis of blood spots for metabolic disorders funneling into more expensive and resource-intensive genetic screening techniques as the case warrants. Proper implementation of this program would result in a wealth of data due to the grassroots-level approach of the RBSK focusing on engaging the existing pool of community health workers.
Although the definition of a rare disease and the focus of this review are based on the prevalence of the disease, estimating the incidence of a disease becomes particularly important in rapidly progressing rare diseases that leave patients with a short life span after diagnosis.
Role of genetics and heterogeneity in rare disease epidemiology
The 100,000 Genomes Project has substantially increased the yield of genomic diagnoses in rare disease patients using genome sequencing. This diagnostic benefit was also seen in patients who had undergone previous genetic testing – Diagnostic yields were 31% in those previously genetically tested and 33% among newly genetically tested. This allowed for immediate medical intervention options to be available for a quarter of those who received a genetic diagnosis[78].
The estimation of the prevalence of rare genetic diseases in India is limited by the lack of a centralized clinical grade (good clinical practice or GCP) registry of patients with rare genetic diseases. The Genomics for Understanding Rare Diseases: India Alliance Network (GUaRDIAN) is an initiative based on the collaboration of 70 institutions utilizing genomics for rare disease research in India to improve “health care planning, implementation, and delivery in the specific area of rare genetic diseases”[79]. Studies conducted by the consortium and others have highlighted the genomic heterogeneity and high prevalence of genetic diseases in the Indian population. The diseases also manifest heterogeneously due to many factors such as subpopulation variations, inbreeding or consanguineous marriage practices, and founder effects and genetic heterogeneity[39,80-83]. There is also heterogeneity in the distribution of the prevalence of rare diseases. Some diseases may be rare in some parts of the world but more prevalent in others, and certain diseases disproportionately affect particular populations. An example is Sickle Cell Disease, which is more prevalent in populations of African, Arabian, and Indian origin. There is considerable heterogeneity within sub-populations as well – the disease is most commonly found among the tribal peoples in central India[84]. Similarly, diseases such as amyotrophic lateral sclerosis[85] or Cystic Fibrosis[86] are considered to predominantly affect people of Caucasian descent. Even though this heterogeneity could be due to challenges and inequitable access to diagnosis resulting in underdiagnosis of these diseases in certain countries, it still raises the question of whether the genetic literature available from other populations can be applied and extrapolated to India and underlines the need to study local populations urgently[16]. Studies that explore the natural history of disease, as well as genotypic and phenotypic expressions at multiple sites across the country, are required.
Emerging genomic techniques such as whole-exome sequencing (WES), and next-generation sequencing (NGS), can increase diagnostic yield and reduce the time to diagnosis while reducing costs. Information regarding the incidence and the carrier frequency of particular disorders can facilitate planning services for genetic counseling for affected families and in areas where the disease is prevalent. Earlier diagnosis of these disorders permits timely intervention in cases where treatment may be possible. Genetic counseling and prenatal diagnosis in affected families still form the basic strategy for lowering the number of individuals with rare disorders in India[11].
A particular challenge to genetic diseases is that phenotypic expression and genetic mutation do not correlate clearly. For example, affected siblings and twins can show different manifestations and severity in Gaucher disease and Adrenoleukodystrophy[87,88]. This is also complicated by de novo mutations frequently arising for genetic diseases[47,50]. In SCN8A epilepsies, patient-reported mutations (n = 140) surpass mutations reported in the published literature[89].
Skewed distribution of rare disease research and funding
Out of the 9,408 clinical entities (groups of diseases, disorders, and sub-types) in the Orphanet database, epidemiological data are available for only 63%[70,90]. There are various reasons for the paucity and skewed distribution of rare disease literature. For instance, in the European Union, rare disease research and funding are prioritized, thereby influencing the quantity and quality of publications[91]. National health expenditure depends on countries’ wealth and resources. Healthcare infrastructure, scientific expertise, and research capacity also vary between different countries, as do their public health priorities. Developed countries have effectively handled leading killers of populations such as infectious diseases, maternal and neonatal morbidity, and others, while these are a major focus of public health measures in LMICs. It is also well known that diseases that are more prevalent in the Global North receive more funding for research. This skews the relationship between diseases prevalent in LMICs and the funding they receive from global agencies. Disease severity and life expectancy can influence prevalence, thereby limiting research opportunities[92]. The peer review process, methodological challenges, and publication bias for positive results also influence the rare disease research that gets published[74,93].
Literature search for epidemiological studies is limited by terminology, language, and dispersion of literature over the decades[70]. Data are spread over and fragmented over a number of different databases – Orphanet has 9,408 rare diseases, of which 4,833 are within the Unified Medical Language System (UMLS), 1753 within the Medical Subject Headings (MeSH®), and 4491 within the Online Mendelian Inheritance in Man databases (OMIM). MeSH® terms for rare diseases have become common only around 2010[94].
Use of ChatGPT, AI/ML methods, and real-world data
A comprehensive assessment of ChatGPT and AI/ML methods is out of the scope of this article. However, we did ask ChatGPT questions such as, “How many patients exist with Pemphigus Vulgaris in the world?”, or “What is the prevalence of Pemphigus or Pemphigoid?”. The estimates received as of 23 April 2023 were similar to those reported in Table 3 with systematic review cited. However, when we asked ChatGPT the question, “What method do we use for estimating the prevalence of a rare genetic disease?”, it returned the response, “Oops Please try again later”[95]. This indicates that the usefulness of ChatGPT and AI tools may be currently limited to quickly reporting the data and information from published literature but fall short in offering novel methodologies for estimating the prevalence of rare diseases.
The US FDA has issued recent guidelines for the use of real-world data including patient-reported outcomes, medical claims, prescription drug databases, electronic health records, and product registries[96]. Patient groups and researchers are also exploring social listening methods to gather real-world data. These offer new opportunities, although scientific validation and evidence may be necessary for specific use.
LIMITATIONS
The quality of articles included in this review has not been evaluated on any scale. However, the articles are from peer-reviewed indexed databases, thus ensuring fair quality. Additionally, the search terms and search were not exhaustive and were carried out on PubMed and Google Scholar due to the broad and extensive nature of the review. The flowchart for the proposed decision tree is non-exhaustive and is based mainly on the studies we have analyzed for this study. The flowchart does not take into account race and ethnicity-based disease occurrence. Even though the scope of the review is limited to ten rare diseases, the learnings from the analysis can be applied to other diseases.
CONCLUSION
Our analysis of the prevalence of a subset of 10 rare diseases in India highlighted the lack of data regarding the epidemiology of rare diseases in India. Existing methodologies for epidemiological studies may not work in resource-constrained countries such as India. There is a stark difference in research capacity between countries belonging to different income levels; this gradient is even more evident for rare disease research due to multiple competing research priorities and the more demanding resource requirement for rare disease research. This has led to a lack of data on the prevalence and incidence of rare diseases in these regions. Based on our analysis, a decision tree has been proposed that can be used as an aid in selecting the methods for studying the prevalence of a rare disease in India and other LMICs.
Global collaborations such as those facilitated by organizations such as the Indo US Organization for Rare Diseases (IndoUSrare) can help bridge the gaps across borders by combining complementary resources and strengths to minimize global inequities in rare disease research[97]. More work needs to be done in capacity building through exchange programs, skill transfer, and mentorship programs to set up rare disease registries and conduct genetic studies. Rare disease research needs to be synchronized globally to arrive at optimized methods that are also viable in resource-constrained settings. Developing newer methods and decentralized technologies for ensuring patient representation in all stages of rare disease research is critical – Empowering patient groups to create and maintain patient registry and natural history studies where data are stewarded and shared by a qualified data sharing committee help estimate prevalence and a better understanding of the disease progression. Such data have the potential to be the basis for clinical trials and the creation of successful public health policies to respond to rare diseases.
